FDA Pharmacovigilance Inspections 2026 | Beyond Documentation
FDA inspections now go beyond SOPs. See real 2026 trends, warning letter insights, and how to stay inspection-ready.

For a long time, pharmacovigilance compliance was treated like a checklist. If the SOPs were written, reports were submitted, and everything looked structured, teams felt ready.
That approach no longer works.
Today, FDA pharmacovigilance inspections are built around a much sharper question: Does your system actually work, or does it just look compliant?
Documentation is still required. But it no longer proves compliance. It only opens the door for deeper scrutiny.
What Has Actually Changed in FDA Pharmacovigilance Inspections
The regulations themselves haven't changed.
What has changed is how the FDA interprets and inspects compliance.
Inspectors are no longer reviewing documents in isolation. They are following the life of a case through your system. They want to understand how information flows, where decisions are made, and whether those decisions make sense.
A system can look clean on paper and still fail if execution breaks under real conditions.
Pharmacovigilance Compliance Rules vs Real Inspection Expectations
Pharmacovigilance obligations in the U.S. are still governed by:
- 21 CFR 314.80
- 21 CFR 600.80
These define how adverse events must be reported.
| Report Type | Timeline |
|---|---|
| 15-day alert (serious + unexpected) | 15 days |
| Follow-up reports | 15 days |
| Quarterly reports | 30 days |
| Annual reports | 60 days |
These timelines are clearly outlined in FDA guidance available on FDA.gov.
But here's the shift in mindset:
A missed timeline is no longer treated as a delay. It is treated as evidence of a pharmacovigilance system failure.
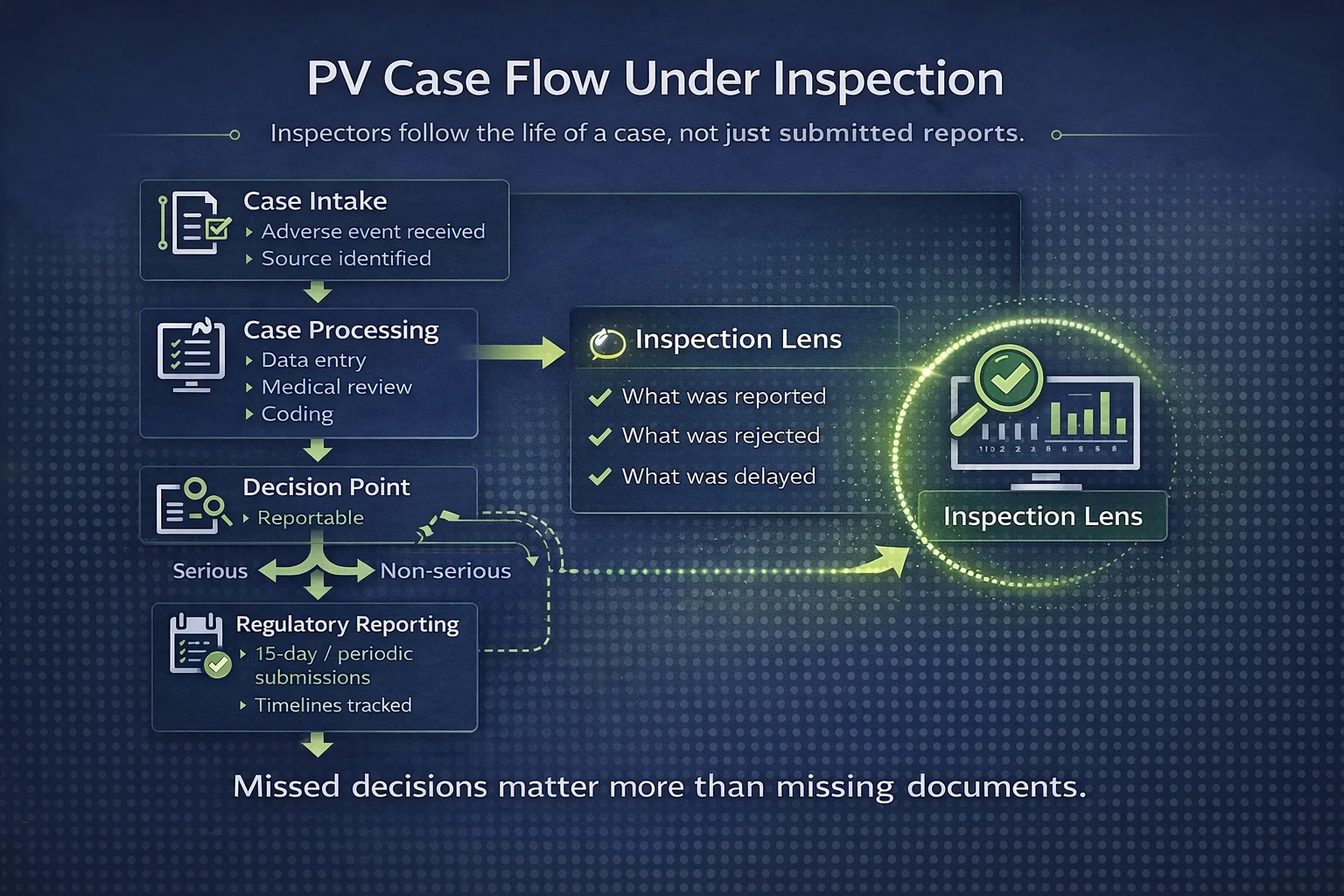
System Failure vs Documentation Compliance
In early 2026, the FDA cited a major pharmaceutical company for excluding serious adverse events based on causality. Their internal logic filtered out events marked as "not related."
From a business perspective, that may seem reasonable. From a regulatory perspective, it is incorrect.
Under U.S. requirements, serious and unexpected events must be reported regardless of causality.
The real issue was not just missed reports. It was this: their pharmacovigilance system was built on the wrong assumption. That's what turned a process gap into a regulatory finding.
Inspectors Now Look at What You Didn't Report in Pharmacovigilance Systems
One of the most important shifts in FDA pharmacovigilance inspections is subtle but critical.
Inspectors don't just review what you submitted. They review what you chose not to submit.
They go into:
- Cancelled cases
- Invalid reports
- Rejected entries
If valid safety data was filtered out, it becomes a serious concern. This directly affects signal detection and broader regulatory decisions.
Data Integrity in Pharmacovigilance Compliance
Data integrity is no longer seen as a backend or IT topic. It is now a core inspection pillar in pharmacovigilance compliance.
Inspectors assess whether your system follows ALCOA+ principles in real practice, not just in documentation. They check if entries are traceable, timestamps make sense, and changes are justified.
Audit trails have become one of the most powerful inspection tools. If data appears to be modified just before an inspection, or if there are unexplained gaps, it immediately raises questions about reliability.
And once data integrity is questioned, the entire PV system comes under scrutiny.
Signal Detection Expectations in FDA Pharmacovigilance Inspections
Pharmacovigilance today is expected to be proactive. It is no longer enough to process incoming cases and wait for patterns to appear.
Inspectors expect to see a defined methodology for identifying risks early. This includes statistical approaches like PRR and ROR, and increasingly, Bayesian methods such as MGPS or EBGM. Real-world data is also becoming part of this expectation.
The underlying question is simple: Are you actively identifying risks, or just recording them?

Vendor Oversight in Pharmacovigilance Compliance
Outsourcing has made pharmacovigilance more scalable, but also more complex.
The FDA's position remains clear: You can outsource the work, but not the responsibility.
What inspectors now look for goes beyond contracts. They expect evidence of actual oversight, including:
- Defined SDEA Key Performance Indicators (KPIs)
- Regular reconciliation between clinical and safety databases
- Visibility into vendor audit trails
- Documented follow-ups when issues occur
If oversight exists only on paper, it is treated as a compliance gap.
Organizations often strengthen this through structured frameworks like Training and Upskilling and Medical Writing.
AI-Driven FDA Inspections and Risk-Based Targeting
Inspections today are far more targeted than before.
For-cause inspections, triggered by safety signals or complaints, now make up nearly 25% of all FDA inspections. More importantly, these inspections are 5.6 times more likely to result in serious regulatory outcomes.
Tools like Elsa are accelerating this shift. They analyze:
- Adverse event narratives
- Reporting patterns
- CAPA effectiveness
- Manufacturing deviations
This allows the FDA to identify risks before stepping on-site. In many cases, inspectors already have a working hypothesis before the inspection even begins.
Form 483 Response Expectations in 2026
The way companies respond to observations is now under more scrutiny. The March 2026 draft guidance on Form 483 responses makes this clear.
Responses are now expected to include:
- A structured and detailed format
- Clear root cause analysis
- A defined remediation plan
And importantly: a formal sign-off from an executive who has the authority to allocate resources.
This is a key shift. Accountability in pharmacovigilance compliance is no longer just functional. It is organizational and personal.
Organizations preparing for this stage often rely on GxP Audits and Pharmacovigilance Consulting to strengthen their response framework.
Inspection Readiness in Pharmacovigilance Systems (2026 Standard)
Inspection readiness is no longer about preparing documents before an audit. It is about running a pharmacovigilance system that performs correctly every day.
That means:
- Reporting decisions are aligned with regulations
- Data is clean, traceable, and consistent
- Vendors are actively managed
- Signals are proactively identified
- Leadership is involved and informed
When all of this is in place, inspection readiness becomes a byproduct, not a project.
Compliance Is No Longer Documents. It Is System Performance
The biggest mistake companies make today is assuming compliance is about having the right documents.
It isn't.
It's about whether your pharmacovigilance system works when something goes wrong. Because that's exactly what the FDA is trying to find out.
If you are evaluating your pharmacovigilance system or preparing for an inspection, you can contact our team or learn more about us.