FDA Warning Letters in Pharmacovigilance | PVCON Consulting
Learn what FDA Warning Letters reveal about pharmacovigilance system controls, adverse event reporting, vendor oversight, PADE compliance, and inspection readiness.

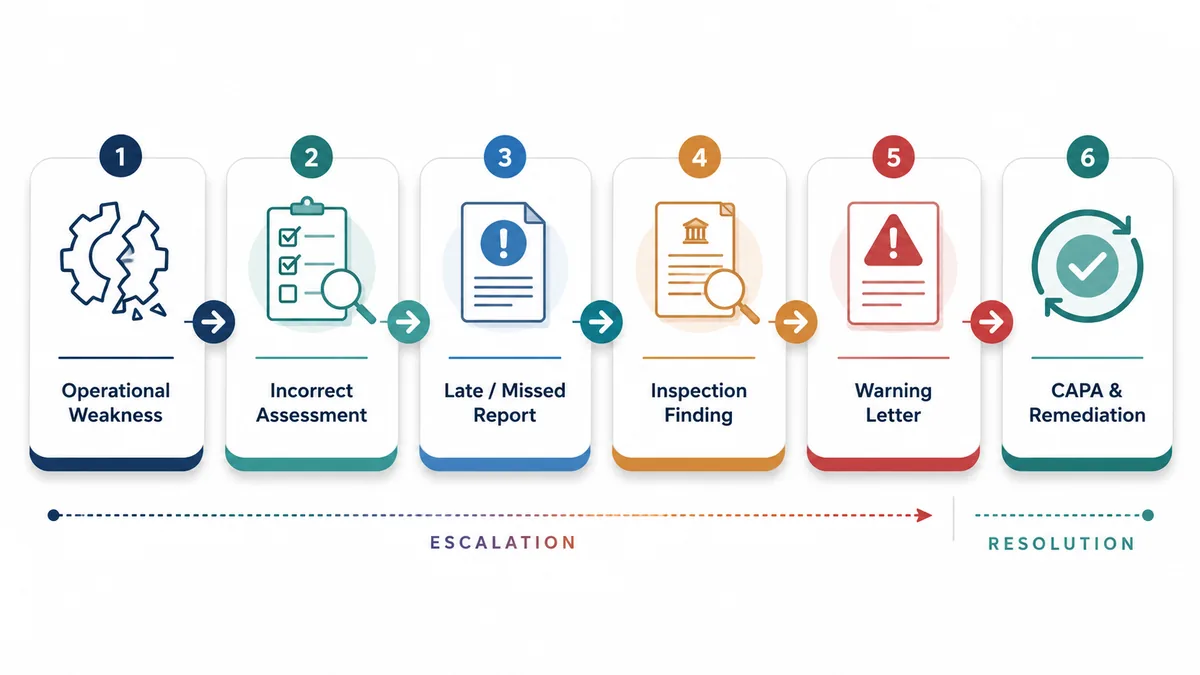
From Compliance to Consequence: What FDA Warning Letters Reveal About Broken PV Systems
FDA Warning Letters are often read as enforcement documents.
For pharmacovigilance teams, they should also be read as system evidence.
A Warning Letter may not point only to an isolated reporting issue. More often, it reveals weaknesses in how safety information is received, assessed, followed up, documented, escalated, and reported.
This distinction matters.
A PV system may have approved SOPs, a validated safety database, trained staff, vendor agreements, and reporting trackers. However, if those controls are not reflected in real safety activity, the system may still be difficult to defend during regulatory review.
FDA's PADE inspection framework and recent Warning Letter examples show how safety reporting concerns may reflect broader limitations in case intake, source review, medical assessment, vendor oversight, and reporting timeline control. A 2026 FDA Warning Letter, for example, stated that investigators reviewed compliance with PADE regulations and identified concerns related to 15-day Alert reporting.
Key Takeaway
FDA Warning Letters reveal more than missed reports.
They may indicate deeper limitations in PV procedures, case validity assessment, seriousness and expectedness evaluation, source document review, medical review timelines, vendor oversight, business partner exchange, and CAPA effectiveness.
The key lesson for MAHs is clear: having PV documentation is not the same as operating a controlled PV system.
Warning Letters Show the System Behind the Finding
FDA Warning Letters should not be reviewed only as a list of cited observations.
They should be reviewed as evidence of how a PV system performed when tested.
A delayed report may reflect late triage. An invalidated case may reflect incorrect source review. An outdated SOP may reflect weak change control. A vendor error may reflect insufficient MAH oversight. A closed CAPA may reflect incomplete effectiveness verification.
This is why Warning Letters should be reviewed by PV operations, quality, medical safety, vendor management, and leadership.
They can indicate where the PV system could not sufficiently demonstrate that its controls operated as intended.
When Written Procedures Do Not Control the Process
Written procedures are central to pharmacovigilance compliance, but they are only effective when they reflect current operations.
In FDA Warning Letter examples involving PADE reporting, the agency has cited violations related to written procedures for the surveillance, receipt, evaluation, and reporting of postmarketing adverse drug experiences.
FDA also noted issues such as unclear responsibilities, obsolete references, insufficient evaluation of seriousness and expectedness, missing procedures for product complaints involving ADEs, and lack of safety information exchange procedures with business partners.
The inspection implication is clear.
An SOP that does not reflect current vendors, forms, responsibilities, complaint pathways, or partner exchanges is not a reliable control.
It may exist as a controlled document, but it may not adequately govern the live PV system.
Organizations reviewing these risks may consider PV Quality Management System support to assess whether procedures, workflows, responsibilities, and evidence trails remain aligned with current pharmacovigilance operations.
Minimum Criteria Can Become a Consequence Point
One of the strongest lessons from FDA Warning Letters is that case intake decisions matter.
A report may fail to move forward because it is incorrectly assessed as invalid. In practice, this can happen when minimum criteria are applied too narrowly or source documents are not reviewed with sufficient care.
A 2026 FDA Warning Letter to a global pharmaceutical company provides a recent example. FDA raised concerns around reports that were invalidated due to alleged missing patient identifiers, while FDA inspection review found patient identifiers in source documents.
This is not only a documentation issue.
It reflects a potential misapplication of minimum criteria, source review weakness, and case intake control concern.
Minimum criteria should support valid case identification. They should not become a barrier that prevents safety information from entering the appropriate PV workflow.
Causality Should Not Become an Intake Filter
Another important issue from the same 2026 FDA Warning Letter involved causality filtering.
FDA identified concerns where cases were rejected or closed because the reporter stated that the event was not related to the product, or because causality was considered excluded.
For PV systems, this is a critical distinction.
Causality assessment may be part of medical review and reportability evaluation, but it should not prevent potentially valid safety information from being received, assessed, and processed according to applicable requirements.
When causality becomes an intake barrier, serious and unexpected events may fail to reach the reporting workflow within required timelines.
Organizations may consider Pharmacovigilance Consulting to review case intake logic, reportability assessment, and workflow alignment across local and global PV operations.
Vendor Oversight Remains MAH Accountability
Many PV systems rely on external vendors, call centers, distributors, affiliates, or business partners to receive and process safety information.
FDA Warning Letters show that delegation does not reduce MAH accountability.
Changing a vendor does not automatically correct a system weakness. If the same invalidation logic, source review limitation, escalation delay, or reconciliation concern continues under a new vendor, the underlying issue remains unresolved.
Vendor oversight should include more than contracts and routine meetings. It should test whether safety data intake, validity assessment, case transfer, follow-up, escalation, and reporting timelines are operating as intended.
A strong vendor oversight model should be evidence-based.
Organizations may consider PV Audits and GxP Audits to assess whether delegated activities are controlled, documented, and inspection-ready.
What MAHs Should Test Before FDA Does
Warning Letters show that PV inspection readiness must be tested through real process evidence, not only document review.
| PV Control Area | What Should Be Tested |
|---|---|
| Case intake | Are all sources of safety information captured and routed correctly? |
| Minimum criteria | Are validity decisions supported by source document review? |
| Medical review | Are serious cases reviewed within controlled timelines? |
| Vendor oversight | Are vendor errors trended, escalated, and corrected? |
| CAPA effectiveness | Does evidence show that the issue was resolved and did not recur? |
This type of review helps move the organization from reactive remediation to proactive inspection preparedness.
It also strengthens confidence that PV documentation reflects real system performance.
As FDA modernizes adverse event reporting through AEMS and the transition toward E2B(R3), manual intake decisions, delayed reconciliation, and weak case documentation will become harder to defend. PV systems should be able to support structured, traceable, and timely safety reporting across both internal and outsourced workflows.
Risk Communication Is Also Safety Governance
Although many Warning Letters focus on case processing and PADE reporting, FDA enforcement also shows that safety governance extends into how risk information is communicated externally.
Promotional materials, risk statements, labeling alignment, and corrective communications can all become part of broader safety governance when risk information is incomplete, minimized, or not presented appropriately.
In a 2025 Warning Letter to a large pharmaceutical organization, FDA raised concerns around risk presentation for prescription products, including boxed warning and serious risk information.
For PV leaders, this reinforces a wider point: safety information must be controlled across the product lifecycle, from adverse event intake to public risk communication.
Organizations can use Regulatory Intelligence to monitor evolving expectations, enforcement trends, and inspection themes across pharmacovigilance and safety communication.
FDA Warning Letters reveal the difference between having PV compliance documents and operating a controlled PV system.
They show how weaknesses in intake, assessment, follow-up, oversight, and reporting timelines can move from internal process issues to formal regulatory consequences.
For MAHs, the practical question is not only, "Are our SOPs approved?"
It is, "Can our PV system prove that safety information was received, assessed, followed up, and reported correctly?"
This is where a structured review of PV governance, written procedures, vendor oversight, reporting workflows, CAPA effectiveness, and inspection readiness becomes essential.
How PVCON Consulting Supports FDA Inspection Readiness
PVCON Consulting supports pharmaceutical, biotechnology, CRO, and medical device organizations through specialized services including GxP Audits, PV Audits, GCP Audits, Other GxP Audits, Pharmacovigilance Consulting, PV Quality Management System support, PvOIC services, Regulatory Intelligence, Medical Writing, Aggregate Report Writing, Clinical Safety Documents, RMP and REMS Writing, PSMF Management, and Training & Upskilling initiatives such as Training Matrix, Regulatory Compliance Training, PV Boot Camp, and Customized Learnings.
Our expertise helps organizations strengthen drug safety operations, improve inspection and audit readiness, and keep PSMF documentation compliant, accurate, and aligned with real-world PV system practices and regulatory expectations.
If you are reviewing your PADE compliance or preparing for an FDA inspection, you can contact our team or learn more about us.